




Prof. William L. Jorgensen (Yale University, New Haven, CT, USA)
Efficient Drug Lead Optimization Guided by Free-Energy Calculations
Abstract
Drug development is being pursued through computer-aided structure-based design. For de novo lead generation, the BOMB program builds combinatorial libraries in a protein binding site using a selected core and substituents, and QikProp is applied to filter all designed molecules to ensure that they have drug-like properties. Monte Carlo/free-energy perturbation simulations are then executed to refine the predictions for the best scoring leads including ca. 1000 explicit water molecules and extensive sampling for the protein and ligand.
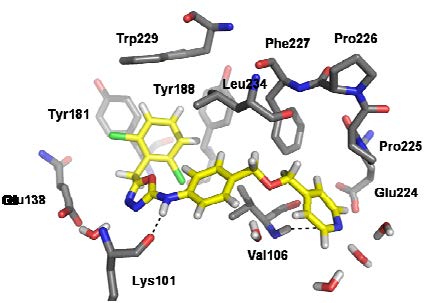
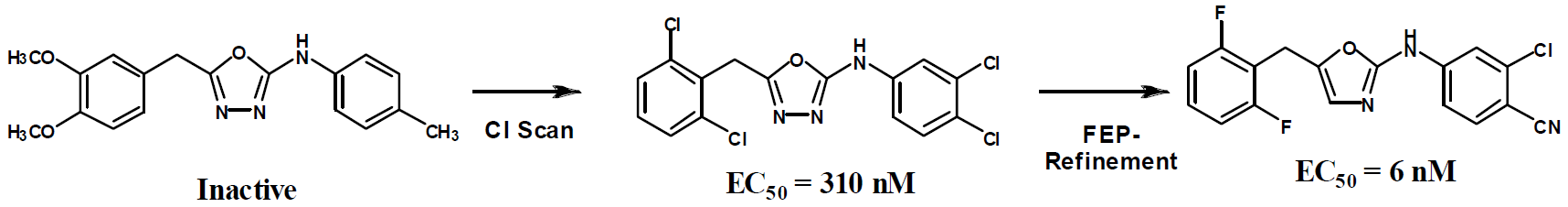
FEP calculations for optimization of substituents on an aromatic ring and for choice of heterocycles are now common. Alternatively, docking with Glide is performed with the ZINC database to provide leads, which are then optimized via the FEP-guided route. Successful application has been achieved for HIV reverse transcriptase, FGFR1 kinase, and macrophage migration inhibitory factor (MIF); micromolar leads have been rapidly advanced to extraordinarily potent inhibitors.
References
- Jorgensen, W. L., Acc. Chem. Res. 2009, 42, 724-733, Efficient Drug Lead Discovery and Optimization.
- Cournia, Z.; Leng, L.; Gandavadi, S.; Du, X.; Bucala, R.; Jorgensen, W. L., J. Med. Chem. 2009, 52, 416-424, Discovery of Human Macrophage Migration Inhibitory Factor (MIF)-CD74 Antagonists via Virtual Screening.
- Leung, C. S.; Zeevaart, J. G.; Domaoal, R. A.; Bollini, M.; Thakur, V. V.; Spasov, K.; Anderson, K. S.; Jorgensen, W. L., Bioorg. Med. Chem. Lett. 2010, 20, 2485-2488, Eastern Extension of Azoles as Non-nucleoside Inhibitors of HIV-1 Reverse Transcriptase: Cyano Group Alternatives.




