The study's findings provide insights into the surprising evolution of proteins
A team of researchers from the Faculty of Science at Charles University, BIOCEV and IOCB Prague (Czech Republic), Johns Hopkins University (USA) and ELSI (Japan) has discovered why modern proteins use a quasi-universal repertoire of 20 canonical amino acids (AAs). The study, published in the journal JACS, shows that foldability was likely a critical factor in the selection of the canonical alphabet.
Previous evidence suggests that ancient proteins relied on a limited alphabet of 10 'early' AAs, and that the 10 'late' AAs were products of biosynthetic pathways. However, many non-proteinogenic AAs were prebiotically available, which begs the question: why do we have the current modern amino acid alphabet and would proteins be able to fold into globular structures as well if different amino acids comprised the genetic code?
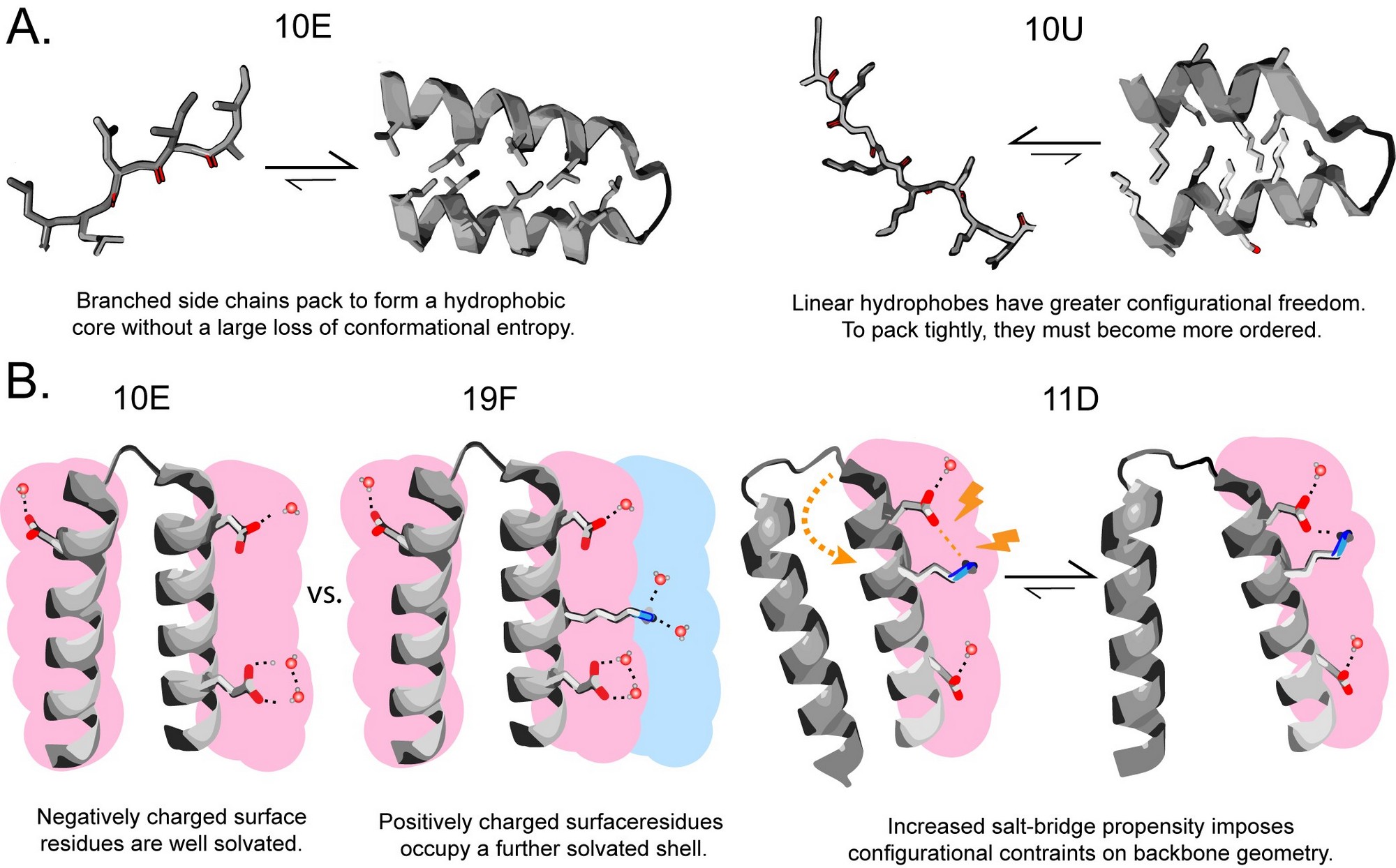
To answer this question, the team experimentally evaluated the solubility and secondary structure propensities of several prebiotically relevant amino acids in the context of synthetic combinatorial 25-mer peptide libraries. The most prebiotically abundant linear aliphatic and basic residues were incorporated along with or in place of other early amino acids to explore these alternative sequence spaces.

The results show that unbranched aliphatic amino acids were purged from the proteinogenic alphabet despite their high prebiotic abundance because they generate polypeptides that are over-solubilized and have low packing efficiency. Surprisingly, the inclusion of a short-chain basic amino acid also decreases polypeptides' secondary structure potential, for which the team suggests a biophysical model.
The study's findings support the view that despite lacking basic residues, the early canonical alphabet was remarkably adaptive at supporting protein folding and explain why basic residues were only incorporated at a later stage of protein evolution.
"We are excited to have uncovered some of the reasons why the protein alphabet has evolved the way we know it now," said the study's lead authors Stephen D. Fried and Klara Hlouchova. "Our findings show that foldability played a critical role in the selection of the canonical alphabet and explain why some prebiotically available amino acids were purged from the proteinogenic alphabet."
Read the paper: Makarov M, Sanchez Rocha AC, Krystufek R, Cherepashuk I, Dzmitruk V, Charnavets T, Faustino AM, Lebl M, Fujishima K, Fried SD, and Hlouchova K. (2023). Early Selection of the Amino Acid Alphabet Was Adaptively Shaped by Biophysical Constraints of Foldability. J. Am. Chem. Soc. https://doi.org/10.1021/jacs.2c12987