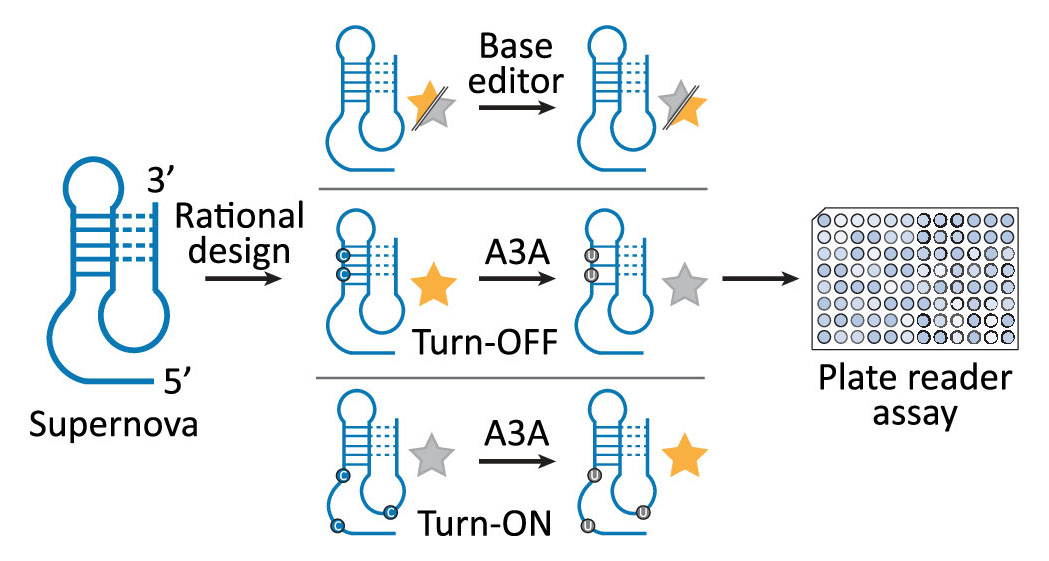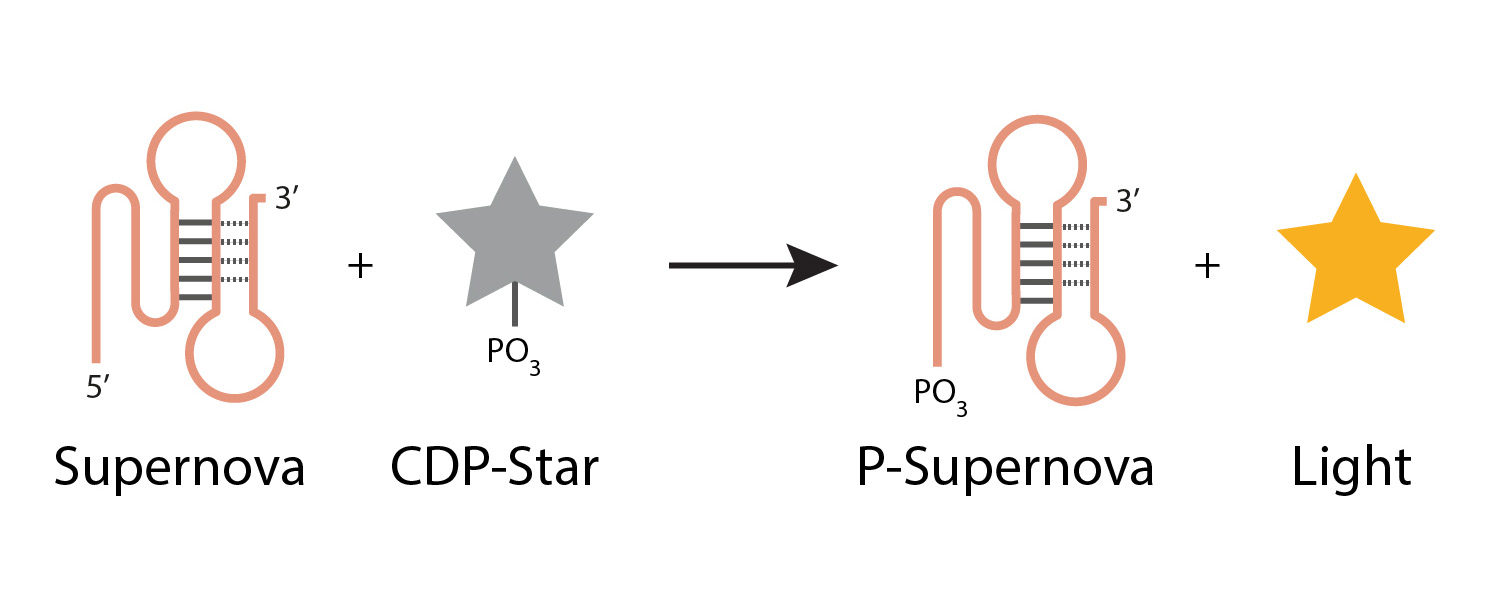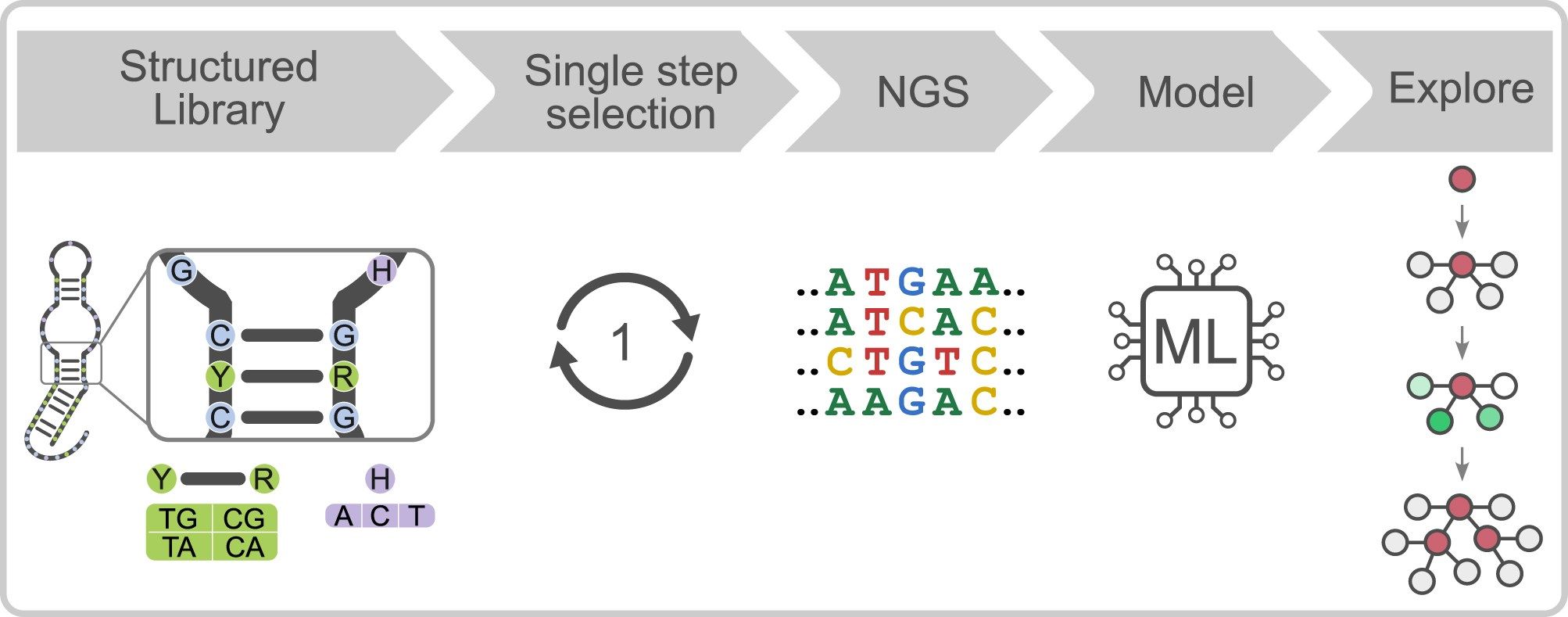
A new paper from the group of Ed Curtis at IOCB Prague in collaboration with the Pluskal group introduces a structure-guided way to explore nucleic acid sequence space that goes far beyond traditional random mutagenesis. By combining rational library design, single-round selections, high-throughput sequencing, and machine learning, the authors rapidly identified catalytically active variants of a fluorescent deoxyribozyme and elucidated predictive rules linking sequence to function. The study was published in the journal Nucleic Acids Research.
Understanding how sequence determines function remains a central challenge in nucleic acid engineering. Conventional strategies typically rely on libraries generated by random mutagenesis of a single variant of a motif, which provide dense coverage close to the original sequence but little information about more distant variants. This limitation makes it difficult to uncover more general principles governing complex functional motifs such as catalytic nucleic acids.

In a new study led by Edward A. Curtis, with Jaroslav Kurfürst and Martin Volek as the first authors, the researchers introduce a fundamentally different strategy for exploring sequence space that combines structure-informed library design, single-round selections, high-throughput sequencing, and machine learning. Instead of randomly mutating a single sequence, the team constructed a library composed of sequences that already satisfy multiple structural and functional constraints of a target motif. This approach was tested on Aurora, a fluorescent deoxyribozyme previously discovered in the group.
The results indicate that this approach is a more efficient way to explore sequence space than standard approaches. A secondary structure library based on Aurora yielded approximately 40 times more unique catalytic sequences than a conventional randomly mutagenized library, and active variants could be rapidly identified in a single round of selection. High-throughput sequencing data were then used to train machine learning models, which successfully identified rules relating sequence to activity. Remarkably, these models could accurately predict read numbers and identify the most active variants using only small subsets of sequences for training.
By integrating rational library design with data-driven analysis, the study demonstrates a scalable way to explore nucleic acid sequence space more rapidly and comprehensively than standard methods allow. Beyond fluorescent deoxyribozymes, this framework provides a general blueprint for accelerating the discovery and optimization of functional nucleic acids, while simultaneously extracting predictive rules that link sequence to function.
Read the paper
- Kurfürst, J.; Volek, M.; Samusevich, R.; Pluskal, T.; Curtis, E. A. Using Structured Libraries, Selection, and Machine Learning to Rapidly Explore the Sequence Space of a Fluorescent Deoxyribozyme. Nucleic Acids Res. 2025, 53 (22). https://doi.org/10.1093/nar/gkaf1348